

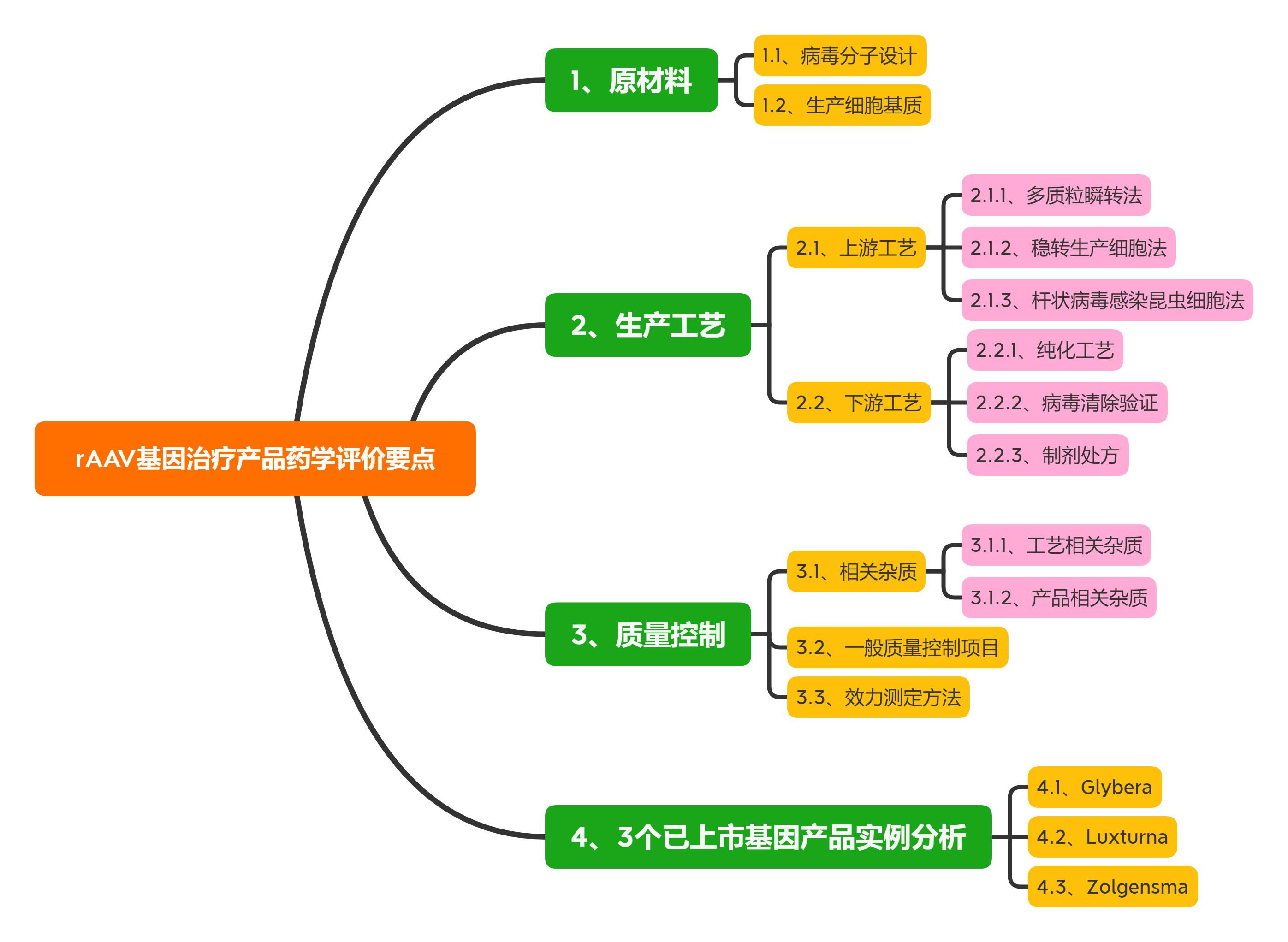
重組腺相關病毒(recombinant adeno-associated virus, rAAV) 載體與其他病毒載體相比, 具備感染能力強、可持續表達目的基因、無致病性和非基因組整合等優點, 已成為體內基因治療的主要病毒載體。
本文總結了rAAV基因治療產品的最新研究進展, 從原材料、生產工藝及質量控制等方面對此類產品的藥學評價考慮要點展開討論, 以期促進此類產品的臨床轉化與應用。同時對3個國外已上市產品的藥學評價考慮要點及常見問題進行討論。
據不完全統計, 目前至少有238 個rAAV基因治療產品正在開展臨床試驗, 3 個rAAV基因治療制品已在歐美注冊上市(表1)。
表1 rAAV基因治療產品的臨床應用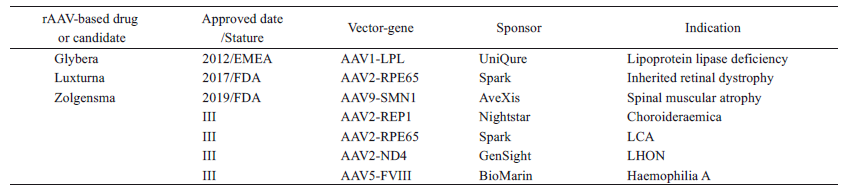
rAAV的載體設計應基于臨床有效性和安全性, 一般為靶向特定組織或細胞, 刪除與毒力、致病性或復制能力相關的基因, 以確保制品的安全性。同時, 設計應考慮載體基因組的大小、包裝效率和表達效率, 并盡可能減少與體內相關病毒的同源性, 以避免形成復制型病毒。構建病毒的質粒應考慮抗生素抗性基因引入的風險, 一般不得使用氨芐青霉素抗性基因。
目前產業界用于rAAV生產的細胞基質既有哺乳動物細胞(HEK293、BHK)、人腫瘤細胞系(HeLa、A549),也包括昆蟲細胞系(SF9) 等。
包裝細胞應進行充分的鑒定并建庫。鑒定項目一般包括鑒別、純度、基因型/表型、成瘤性/致瘤性、遺傳穩定性和引入序列等。除常規無菌、真菌和支原體外, 尤其要關注細胞基質的種屬特異性病毒。如: HEK293 細胞應檢定CMV、HIV-1/2、HILV-1/2、HH-V6/8、JV virus、BK virus、EBV、parvovirusB19、HBV、HPV和HCV等病毒; SF9 細胞應檢定螺原體、彈狀病毒等昆蟲病毒。
用人腫瘤細胞系作為包裝細胞, 還應充分評估其致瘤性和成瘤性的風險, 以及致瘤病毒的感染情況等, 如: HeLa 細胞為來自人子宮頸癌組織的腫瘤細胞, 已知含有50 個拷貝數的HP
最早的rAAV 生產工藝采用質粒、輔助病毒感染包裝細胞生產。目前采用較多的是三質粒共轉染HEK 293 生產工藝, HEK293 細胞中含有腺病毒(adenovirus, AdV) 的E1a 和E1b 基因, 共轉染轉移質粒(含目的基因和ITR序列)、結構質粒(含rep/cap 基因) 和輔助質粒( 復制病毒基因E2A, E4,VARNA 等), 48~72 h 后即可重組包裝rAAV。
該方法適用于多種血清型rAAV, 發酵產率一般可達每毫升1014 V.G. (vector genome), 一般可滿足早期臨床試驗rAAV用量(<1015 V.G.)。但是, 受貼壁培養方式所限, 多質粒瞬轉工藝較難滿足商業化生產需求(>1016 V.G.)。
構建含有輔助基因rep/cap和目的基因的穩轉HeLa 細胞系, 經腺病毒感染后也可包裝出rAAV 病毒。尤其是采用可懸浮培養的HeLa S3細胞系, 穩轉細胞生產法可直接放大至2 000 L規模。與之類似, 構建含有ICP27 基因的BHK 細胞,經轉染含有輔助基因和目的基因的復制缺陷型d27.1HSV后, 也可高效表達rAAV。
但是, 穩轉細胞法的主要缺點在于, 細胞構建和遺傳穩定性研究較為耗時,且生產過程中使用輔助病毒存在病毒安全性風險。
桿狀病毒具有高度的種屬特異性, 不感染脊椎動物, 能將rAAV基因和輔助功能的反式作用元件轉移至SF9 昆蟲細胞中。因此, 近年也開始應用于rAAV 的大規模生產。該方法的優勢在于桿狀病毒生物安全性好, 感染效率高, 生產工藝便于放大。但質量控制項目中也應考慮桿狀病毒及DNA相關物質殘留。
藥學評價要點: 對于rAAV 上游生產工藝, 藥學評價建議關注關鍵工藝的工藝開發與驗證, 如采用多質粒瞬時轉染或加入輔助病毒等工序中, 應通過工藝開發與驗證說明關鍵工藝參數的控制范圍及中間體驗收標準, 如不同載體配比、轉染試劑用量、輔助病毒與生產細胞的感染復數等。
rAAV 純化工藝一般包括細胞培養液收獲、化學法/物理法裂解細胞、benzonase 酶消化去除核酸物質、多步層析和密度梯度離心、置換制劑處方緩沖液等工序, 其中, 親和層析模擬細胞受體的結合作用捕獲rAAV。如: 硫酸肝素填料可特異性結合rAAV2, AVB瓊脂糖填料可結合1、2、3、5 和8 等多種血清型rAVV[22]。對于空載體的去除, 目前多采用碘克沙醇和氯化銫超速離心法。
值得關注的是, 中國藥典相關總論中明確指出,“除另有證明其合理性外, 不得使用氯化銫-溴化乙錠密度離心法進行基因治療產品的純化”。
如上所述, 采用桿狀病毒-昆蟲細胞或穩轉細胞系(HeLa) 生產工藝時會使用桿狀病毒或輔助病毒(HSV、Ad5 等)。因此, 下游工藝應增加特定病毒去除/滅活工序。
如: 采用表面活性劑TritonX-100 (0.5%, v/v) 和增加病毒過濾工序去除收獲液中殘留桿狀病毒; 采用加入表面活性劑、低pH值滅活等工序去除HSV病毒; 采用離子交換法、短時間加熱(52 ℃、10 min) 和納濾等工序去除腺病毒等。
由于rAAV衣殼蛋白對溫度和pH值變化并不敏感, rAAV 制品的制劑處方開發相對簡單。通常慣用在鹽溶液中加入200 mmol·L-1硫酸鎂或0.001%泊洛沙姆F68 避免產生聚體, 基本可支持長期保存(-65 ℃) 和運輸穩定性。
對于rAAV 下游生產工藝, 藥學評價建議結合產品相關雜質、過程相關雜質的去除效率評估純化工藝合理性與穩健性。對于使用輔助病毒的生產工藝應結合收獲液中病毒含量, 進行病毒去除/滅活工藝驗證,綜合評估病毒的殘留安全性。
rAAV 制品的工藝相關雜質包括病毒包裝與純化工藝中引入的宿主蛋白、宿主DNA、輔助病毒、質粒、血清和氯化銫等組分。由于病毒包裝細胞通常含有致癌基因, 如HEK293 細胞內含有E1A 的腺病毒基因, HeLa 細胞內的E6、E7 致癌基因。因此, 一般對于宿主細胞殘留DNA 含量應小于10 ng/Dose, 且殘留DNA片段應小于200 bp。
rAAV 制品的相關雜質包括未包裝基因的空病毒、包裝錯誤基因(宿主DNA、不完整目的基因、輔助病毒基因等) 的病毒、無感染活性的病毒顆粒、聚體或氧化形式的病毒顆粒等。
這些產品相關雜質不僅不能實現目的基因在靶細胞的表達, 還可在臨床上引起免疫原性或基因毒性。如: 多質粒順轉體系中, 一般空病毒占比可以達到50%~98%。空病毒不具感染能力, 且容易形成病毒聚體和降解, 引起體內免疫反應。因此, 質量研究中應采用A260/A280、透射電鏡、分析性離心和質譜等技術檢測空病毒含量。
復制型腺相關病毒(replication-competent AAV,rcAAV) 是由于同源/非同源重組發生后, rAAV病毒同時包裝rep 和cap/AAP等基因所產生的產品相關雜質。 rcAAV在輔助病毒存在的條件下可以進行復制擴增。rcAAV的檢測一般采用在輔助病毒存在下使用敏感細胞進行擴增, 細胞裂解液經過多次擴增傳代后, 采用Southern blot 和qPCR法測定rep 或cap 基因。如: 已進入臨床試驗的rAAV 制品scAAV2/8-LP1-hFIXco 采用qPCR 法控制rcAAV 含量限度低于1/2.25×106 。目前也有報道稱采用優化后的多質粒瞬轉工藝,rcAAV含量限度可低于1/108 V.G.。
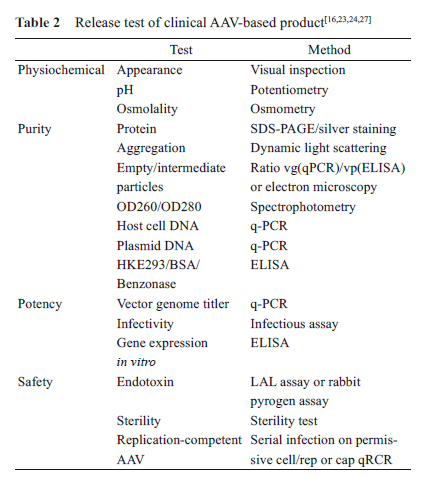
rAAV病毒生產質量控制包括過程控制與終產品放行檢測(表2)。
如: 病毒包裝用質粒應進行鑒別、含量、純度、宿主細胞DNA殘留、轉染效率、細菌內毒素、無菌等質量控制;
病毒收獲液應控制外源因子(無菌、支原體等)、外源病毒、目的病毒等檢測;
rAAV原液與制劑放行項目一般包括外觀、理化性質(pH值、滲透壓)、病毒滴度(物理滴度、感染滴度)、純度(蛋白純度、吸光度比值、宿主DNA殘留、質粒DNA殘留、核酸酶殘留)、效力(目的基因表達、體外活性、體內活性)、安全性 (內毒素、無菌、rcAAV)。
此外, 對于眼部用藥的rAAV 制劑, 內毒素含量應不高于2.0 EU/dose/eye 或0.5 EU/mL, 應按照眼用制劑控制不溶性微粒(USP<789>)、產品的放行檢測應包括配置后產品等。
對于rAAV 制品的效力測定方法, 應體現其病毒物理滴度、感染活性及生物活性。
一般在臨床試驗早期可采用qPCR 法測定病毒基因組, 敏感細胞或靶向細胞測定其感染能力和目標產物表達能力表征產品效力。
關鍵性臨床試驗開展后應進一步開發反映產品作用機制的體外酶活法或體內功能實驗法。如:SPK-RP65 在進入Ⅲ期臨床后, 通過轉染指示細胞(HEK293-LRAT) 后測定RPE65 蛋白酶促反應產物視黃醇含量來計算產品效力。
值得指出的是, 對于qPCR法測定病毒基因組應采用線性DNA繪制標準曲線, 若采用超螺旋DNA作為參照品, 載體基因組含量測定值顯著高于真實值。
目前, 國際上僅有3個rAAV基因治療產品(glybera、luxturna 和zolgensma) 獲批上市, 國內此類產品多處于研發早期階段, 工業界與監管方對于rAAV 基因治療產品的藥學研究與評價均缺乏實踐經驗。下文擬結合國外已上市產品審評報告中披露信息, 對此類產品的藥學評價考慮要點及常見問題進行討論。
Glybera (alipogene tiparvovec, AAV1-LPLS447X)是Amsterdam Molecular Therapeutics 公司開發的攜帶人LPLS447X 基因的rAAV2 型基因治療產品, 臨床上肌肉注射用于治療成人脂蛋白脂肪酶缺陷癥。
本品工藝開發過程中存在重大工藝變更: 第一代生產工藝(AMT-010) 采用HEK293 表達系統, 商業化生產工藝(ATM-011) 則采用桿狀病毒-昆蟲細胞系統表達, 因此開展了藥學和非臨床可比性研究。
下游工藝中采用工藝規模縮小模型對包膜病毒、非包膜病毒去除能力進行了工藝驗證。
質量研究對 3 批連續生產批次制品進行了充分的表征研究, 其中包括: 組分(基因組完整性/大小、蛋白質分析、分子質量、衣殼蛋白); 物理性質(顆粒大小、病毒顆粒糖基化修飾); 一級結構(序列確證、蛋白鑒定); 高級結構(透鏡電鏡、分析超速離心); 生物活性(感染性顆粒、比活、效力) 等。
EMEA審評認為, 由于細胞收獲液中含有大量感染性桿狀病毒, 且下游工藝不能提供足夠的病毒去除效果, 要求申請人補充提供臨床注射桿狀病毒殘留DNA的風險分析報告, 并建議產品放行檢測項目中增加感染性桿狀病毒殘留和rcAVV等檢查項目。
并且, EMEA審評建議產品上市后生產工藝應進一步增加桿狀病毒去除工序(如病毒過濾), 后期應提高雜質(包裝細胞DNA、殘留Rep/Cap 基因、rcAAV、感染性桿狀病毒等殘留) 檢測方法靈敏度。
Luxturna (voretigene neparvovec-rzyl, SPK-RPE65)系美國Spark Therapeutics 公司研發的攜帶人RPE65基因rAAV-2 型基因治療制品, 臨床上視網膜下注射用于治療先天性黑朦癥。
產品采用三質粒共轉染貼壁HEK293 細胞, 滾瓶生產工藝。生產工序包括: 細胞擴增、轉染、培養基置換、收獲培養液、切向流過濾、均質、離子交換色譜、密度梯度離心、制劑緩沖液置換和過濾等;
本品Ⅲ期臨床試驗前曾發生場地、包裝容器等工藝變更, 采用“Side-by-side”法對產品質量的方法進行產品可比性研究。
產品放行檢測項目包括: 理化(外觀、pH值、濃度、可抽取體積)、鑒定(目的基因)、含量(基因組濃度)、效力(目的基因表達、體外活性)、純度和安全性項目(內毒素、顆粒物、無菌) 等, 質量研究中對宿主DNA、質粒DNA、E1A 基因和牛血清蛋白等工藝相關雜質殘留進行控制。
FDA審評認為, 本品上市后應繼續完成包裝細胞HKE293 穩定性及產品實時穩定性等研究。
Zolgensma (onasemnogene abeparvovec, AVXS-101)系AveXis 公司開發的攜帶運動神經元蛋白1 (survival motor neuron, SMN) 基因的rAAV9 型基因治療產品,臨床上靜脈注射治療兒童脊髓肌肉萎縮癥。
本品在工藝開發過程中, 結合目標質量屬性采用風險評估方法確定本品關鍵質量屬性, 采用工藝規模縮小模型確定工藝設計空間并進行工藝驗證;
本品在關鍵性臨床試驗開展前、注冊上市前, 先后發生場地、工藝和制劑處方等變更。
FDA 審評判定工藝變更前后產品純度和VG 單位效力保持一致; 本品的“效力”檢測項中既包括定量檢測靶細胞SMN蛋白表達水平, 也包括體內半定量法檢測小鼠“生存率”。
值得關注的是, 由于本品在? 期臨床試驗開展過程中“含量”分析方法缺乏精確度與準確度, 44 個月后采用更新的方法重新修正臨床給藥劑量。此外, 在本品穩定性實驗中, 觀測到“含量”和“效力”有所下降。
轉自:藥學學報 商圖藥訊